Arzneimittelzulassung

Übersetzungen für alle Arzneimittelzulassungen, inkl. PL und SmPC
Professionell in alle 24 EU-Sprachen bzw. 26 EWR-(EEA-)Sprachen
Die Zulassung von Arzneimitteln ist in Europa strikt reguliert und erfordert in der Regel umfangreiche Fachübersetzungen medizinischer und pharmakologischer Dokumente.
Als spezialisierter Sprachdienstleister hat mpü erstklassige Lösungen für alle Arzneimittel erarbeitet, die sowohl auf internationale Zulassungen zugeschnitten als auch an nationale Regularien angepasst sind.
- Alle Arzneimittel
einschließlich aller Spezialgebiete, z.B.:- Pädiatrische Arzneimittel
- Onkologische Arzneimittel
- Orphan Drugs
- Tierarzneimittel
- etc.
- Generika
- Biosimilars
Arzneimittelzulassungen erfordern Präzision – wir liefern sie. Erfahren Sie mehr über unsere maßgeschneiderten Übersetzungsleistungen.
info@mpue.com
+49 (0)731 954 950
Über 45 Jahre Übersetzungserfahrung für Ihre sichere
Arzneimittelzulassung
Aufgrund der langjährigen Erfahrung und Expertise ist mpü bestens vertraut mit den branchenspezifischen Herausforderungen der Pharma- und Biotechnologie-Industrie.
Mit unserem umfassenden interdisziplinären Leistungsspektrum verfügen wir speziell für Übersetzungen im Rahmen aller Zulassungsverfahren über validierte Prozesse, die allen europäischen sowie nationalen Regularien entsprechen und Behördenakzeptanz sichern:
- Zeitmanagement mit kurzen Lieferfristen
- Sicherheit durch ständige Weiterentwicklung erprobter und bewährter Verfahren
- Durchgängig konsistente Qualität
- Qualitätssicherung durch Übersetzungsvariante nach DIN EN ISO 17100
- Operative Exzellenz für mehr Effizienz und Effektivität

Wir beherrschen 150 Sprachen
Regulatory Labeling – Linguistische Expertise in allen Phasen des Life Cycle
mpü bietet Ihnen ein Komplettpaket aus Lösungen und Leistungen für die Zulassung von Arzneimitteln unter strenger Einhaltung aller Regularien/Vorschriften der Europäischen Arzneimittel-Agentur (EMA) sowie anderer länderspezifischer Behörden.
- Erstellung der englischen Produktinformation (SmPC, PIL, Labeling), auch bekannt als:
- Fachinformation, Zusammenfassung der Merkmale des Arzneimittels, Summary of Product Characteristics (SmPC),
- Packungsbeilage, Gebrauchsinformation, Package Information Leaflet (PIL), Package Leaflet,
- Etikettierung, Etikett(en), Labeling, Text on Inner- and Outer Packaging
- Übersetzung in 26 QC-geprüfte Sprachversionen
- Qualitätssicherung
- Readability User Test
- Linguistic Review
Lassen Sie Ihre Arzneimittelzulassung nicht an linguistischen Hürden scheitern. Wir unterstützen Sie mit erstklassigen Übersetzungen.
info@mpue.com
+49 (0)731 954 950
Übersetzungen im Rahmen von Arzneimittelzulassungen: Vorausschauende Planung – Flexibles Zeitmanagement
mpü garantiert Ihnen sorgfältige vorausschauende Planung des gesamten Übersetzungsprozesses und zeitgerechte Lieferung korrekter und geprüfter nationalsprachlicher Dokumente zur sicheren Einhaltung der Einreichungsfristen.
Timelines des Übersetzungsprozesses der Zulassungsverfahren
Zentrales Zulassungsverfahren (CP): Normales Verfahren
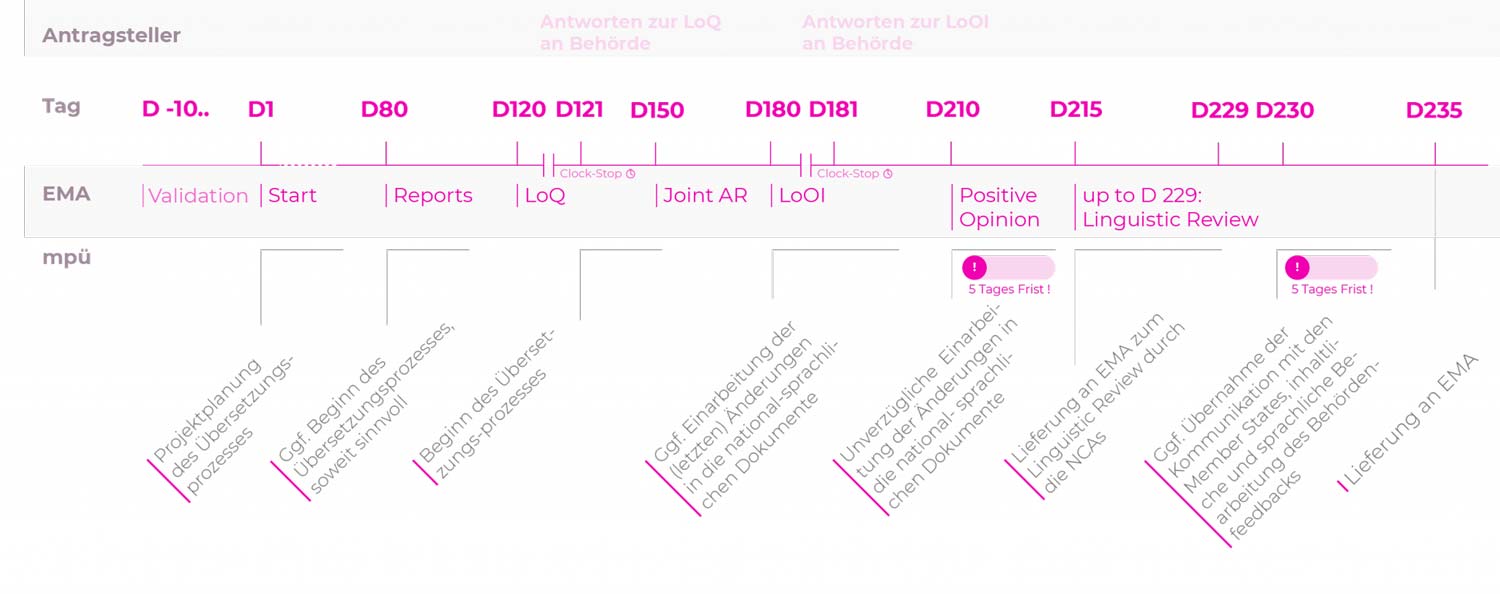
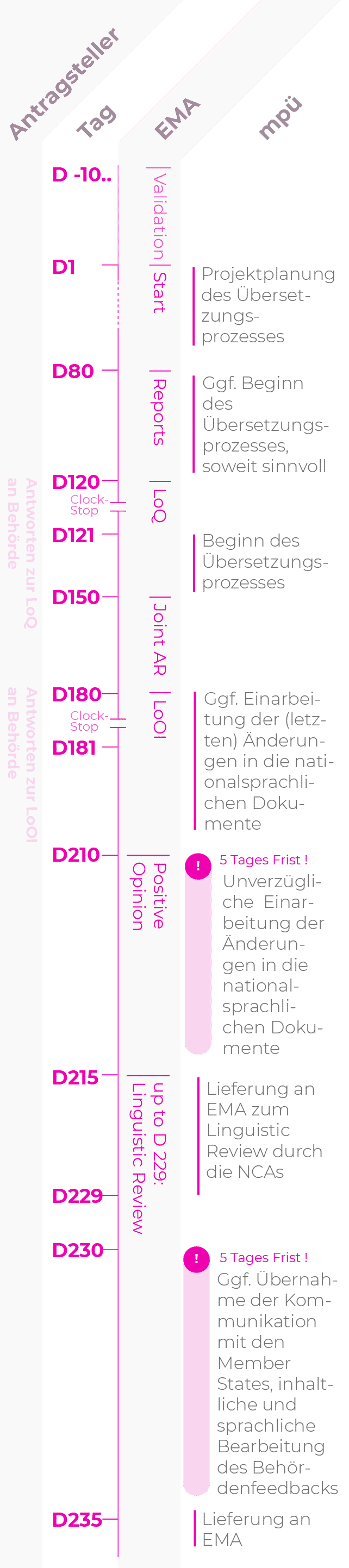
Zentrales Zulassungsverfahren (CP): Normales Verfahren
Die übliche Verfahrensdauer beträgt 180 oder 210 Tage.
| D-10… | Tag der eCTD-Einreichung Der Antragsteller informiert mpü über die Einreichung und stellt die zu übersetzenden Dokumente zur Verfügung. Dies ermöglicht eine verlässliche Prozess- und Ressourcenplanung. |
| D80 | Tag 80, Assessment Report (AR) Die erste Fassung des Assessment Reports (AR) des Rapporteurs/Co-Rapporteurs liegt dem Antragsteller vor. Etwaige Forderungen zur Änderung von SmPC, Package Leaflet oder Labelling müssen jetzt berücksichtigt werden. Übersetzungen unter Einbeziehung der Kommentare im Assessment Report können nun begonnen werden. |
| D120 | Tag 120, List of Questions (LoQ), Beginn CLOCK STOP Falls Forderungen zu Änderungen an Fachinformation (SmPC), Packungsbeilage (PIL) oder Labeling erhoben werden, übermittelt der Antragsteller diese an mpü. |
| D121 | Tag 121, Antworten zur LoQ an die Behörde Mit Einreichung des Response Package übermittelt der Antragsteller die geänderten Dokumente an mpü. |
| D150 | Tag 150, Joint AR |
| D180 | Tag 180, List of Open Issues (LoOI), Beginn CLOCK STOP Falls Forderungen zu Änderungen an SmPC/SPC, Package Leaflet (PL/PIL) oder Labelling erhoben werden, übermittelt der Antragsteller diese an mpü. |
| D181 | Tag 181, Antworten zur LoOI an die Behörde Mit Einreichung der Antworten zur LoOI übermittelt der Antragsteller die geänderten Dokumente an mpü. |
| D210 | Tag 210, Positive Opinion Nach der Positive Opinion sollten nur noch letzte Korrekturen durchgeführt werden. |
| 5-Tages-Frist | |
| D215 | Tag 215, Letzte Korrekturen an die Behörde Lieferung der letzten Korrekturen an die Behörde zum Linguistic Review durch die NCAs. |
| D229 | Tag 215 bis Tag 229, Linguistic Review |
| D230 | Tag 230, Eingang des Ergebnisses des Linguistic Review Der Antragsteller übermittelt notwendige Änderungen an mpü. |
| 5-Tages-Frist | |
| D235 | Tag 235, Lieferung der letzten Überarbeitung an die Behörde |
Zentrales Zulassungsverfahren (CP): Verkürztes Verfahren
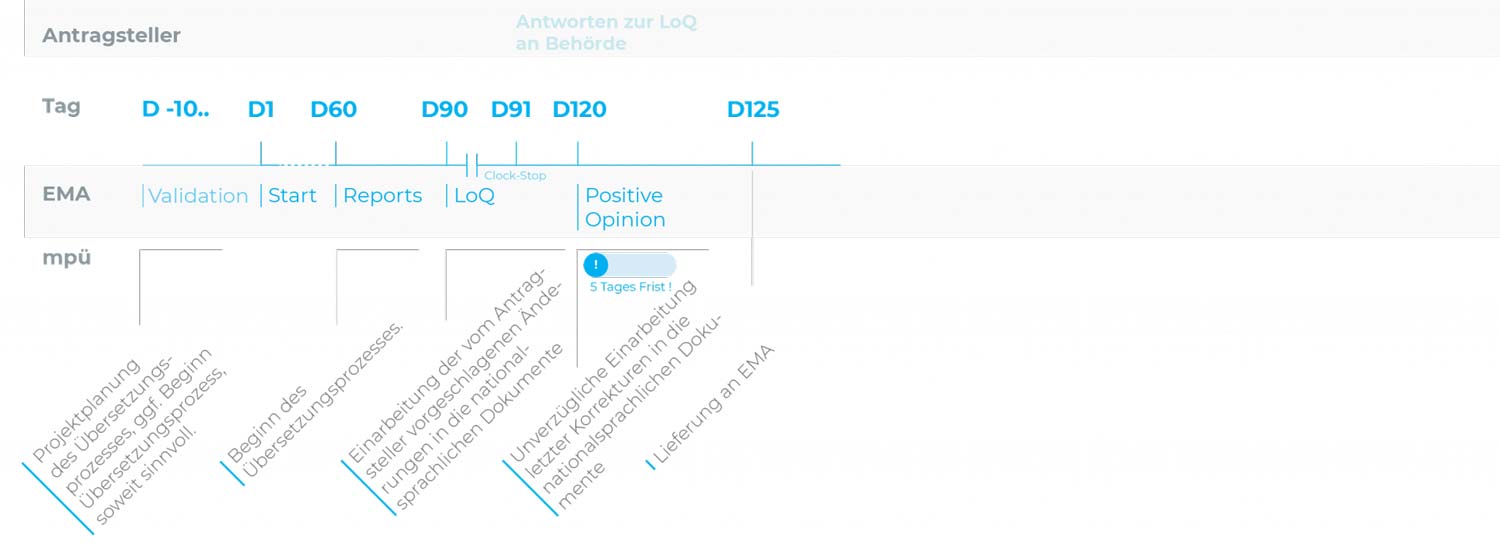
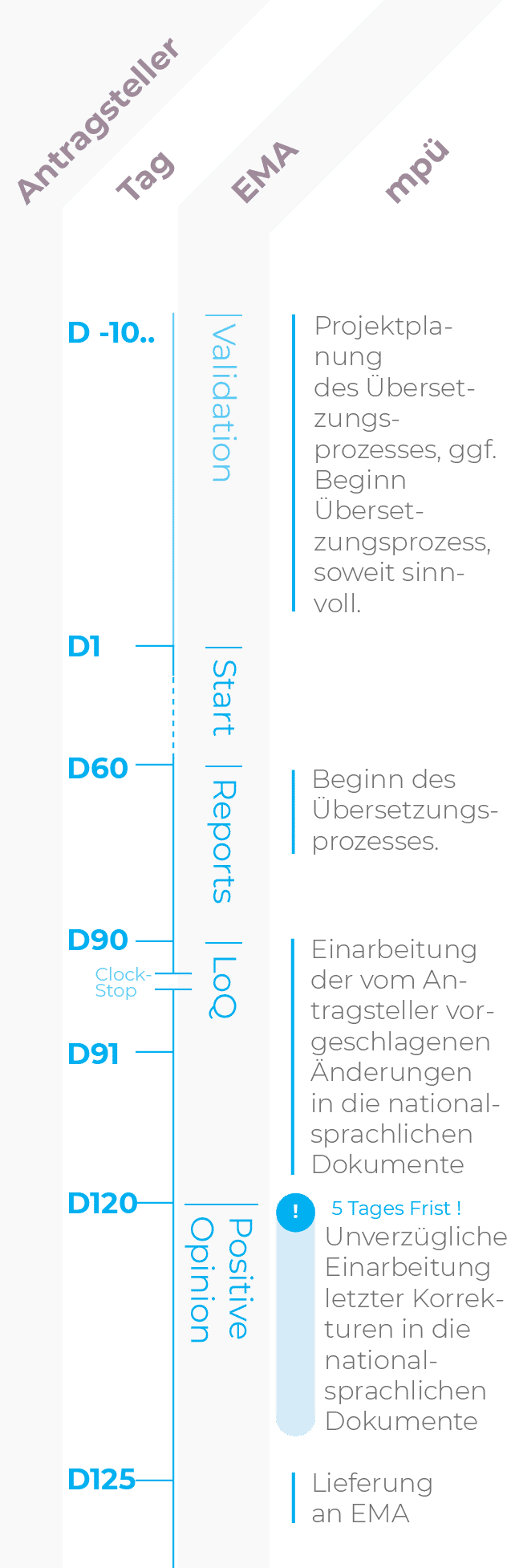
Zentrales Zulassungsverfahren (CP): Verkürztes Verfahren
Die übliche Verfahrensdauer beträgt 90 Tage plus 30 Tage für die Prüfung eines Response Package.
| D-10.. | Tag der Einreichung Der Antragsteller informiert mpü über die Einreichung iund stellt die zu übersetzenden Dokumente zur Verfügung. Dies ermöglicht eine verlässliche Prozess- und Ressourcenplanung. Soweit sinnvoll, kann bereits mit den Übersetzungen begonnen werden. Aufgrund des beschleunigten Verfahrens ist dies ratsam. |
| D60 | Tag 60 Die erste Fassung des Assessment Reports (AR) des Rapporteurs/Co-Rapporteurs liegt dem Antragsteller vor. Etwaige Forderungen zur Änderung von Fachinformation (SmPC), Packungsbeilage (PIL) oder Labeling werden mpü zugeleitet. Spätestens ab Tag 60 sollten die Übersetzungen, unter Einbeziehung der Kommentare im Assessment Report, durchgeführt werden. |
| D90 | Tag 90, Beginn CLOCK STOP Der Antragsteller erhält die List of Questions (LoQ). Falls Forderungen zu Änderungen an SmPC, PIL oder Labeling erhoben werden, übermittelt der Antragsteller diese an mpü. Mit Einreichung des Response Package leitet der Antragsteller die geänderten Dokumente an mpü weiter. |
| D91 | Tag 91, Antworten zur LoQ an die Behörde Mit Einreichung des Response Package übermittelt der Antragsteller die geänderten Dokumente an mpü. |
| D120 | Tag 120 Übermittlung der Positive Opinion an den Antragsteller. Nach der Positive Opinion sollten nur noch letzte Korrekturen durchgeführt werden. |
| 5-Tages-Frist | |
| D125 | Tag 125, Lieferung der letzten Überarbeitung an die Behörde |
Wir sind Ihr linguistischer Partner für die globale Arzneimittelzulassung. Vertrauen Sie auf unsere Expertise für Ihren internationalen Erfolg.
+49 (0)731 954 950
Dezentrales Zulassungsverfahren (DCP)
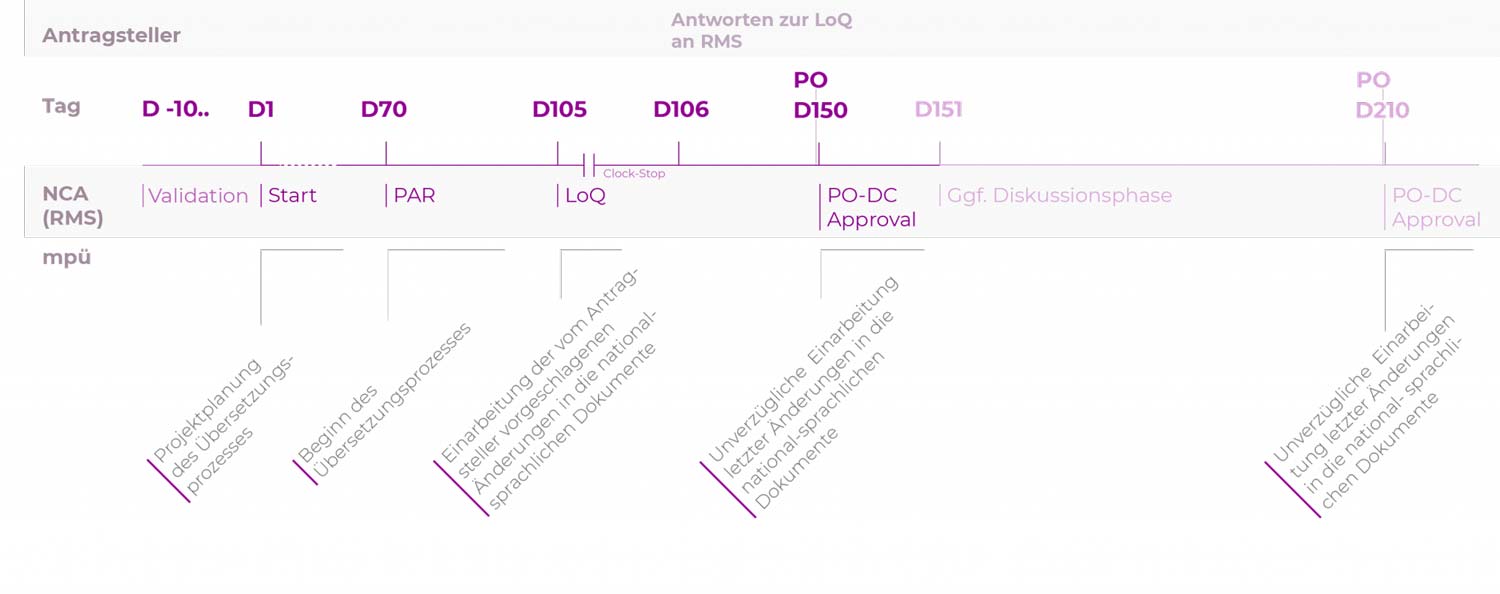
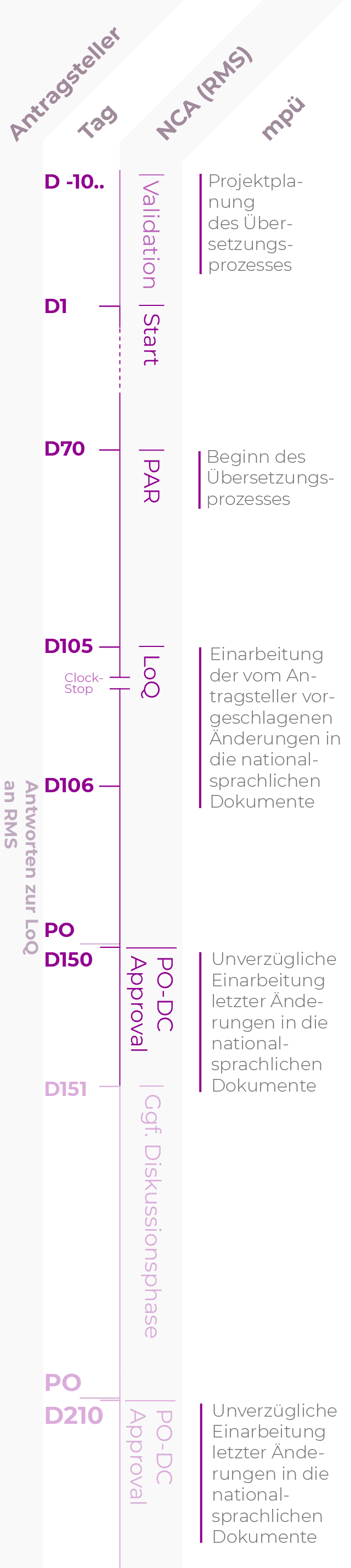
DCP-Verfahren
Die Verfahrensdauer kann 105, 150 oder 210 Tage betragen, abhängig von der Konsensbildung der nationalen Behörden.
| D-10.. | Tag der Einreichung Der Antragsteller informiert mpü über die Einreichung und stellt die zu übersetzenden Dokumente zur Verfügung. Dies ermöglicht eine verlässliche Prozess- und Ressourcenplanung. |
| D70 | Tag 70 Der Reference Member State (RMS) verteilt den „Preliminary Assessment Report“ (PAR) an die beteiligten Mitgliedstaaten („Concerned Member States“, CMS) und den Antragsteller. Spätestens ab Tag 70 sollten die Übersetzungen durchgeführt werden, unter Einbeziehung der Kommentare im PAR. |
| D105 | Tag 105, Beginn CLOCK STOP Dem Antragsteller liegen die Deficiency Statements der Mitgliedstaaten vor. Falls das Response Package Änderungen an der Fachinformation (SmPC), der Packungsbeilage (PIL) oder dem Labeling enthält, werden die geänderten Dokumente mpü mit Einreichung des Response Packages zugeleitet. |
| D150 | Tag 150 Bei Konsens aller MS erteilt der RMS das DC-Approval. Nach dem DC-Approval sollten nur noch letzte Korrekturen durchgeführt werden. Falls noch Punkte ungeklärt sind, informiert der RMS den Antragsteller. |
| Es schließt sich eine weitere Phase der Diskussionen an. | |
| D210 | Tag 210 Üblicherweise wird spätestens am Tag 210 Konsens erreicht und der RMS erteilt das DC-Approval. Nach dem DC-Approval sollten nur noch letzte Korrekturen durchgeführt werden. |
Verfahren gegenseitiger Anerkennung (MRP)
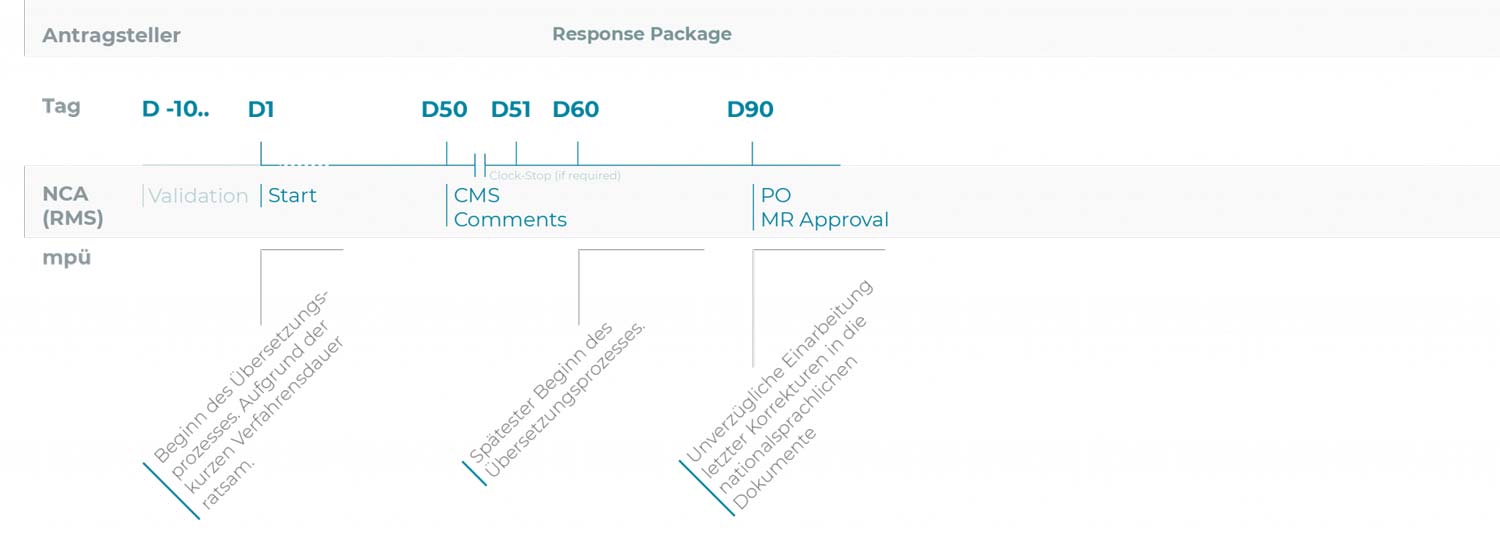
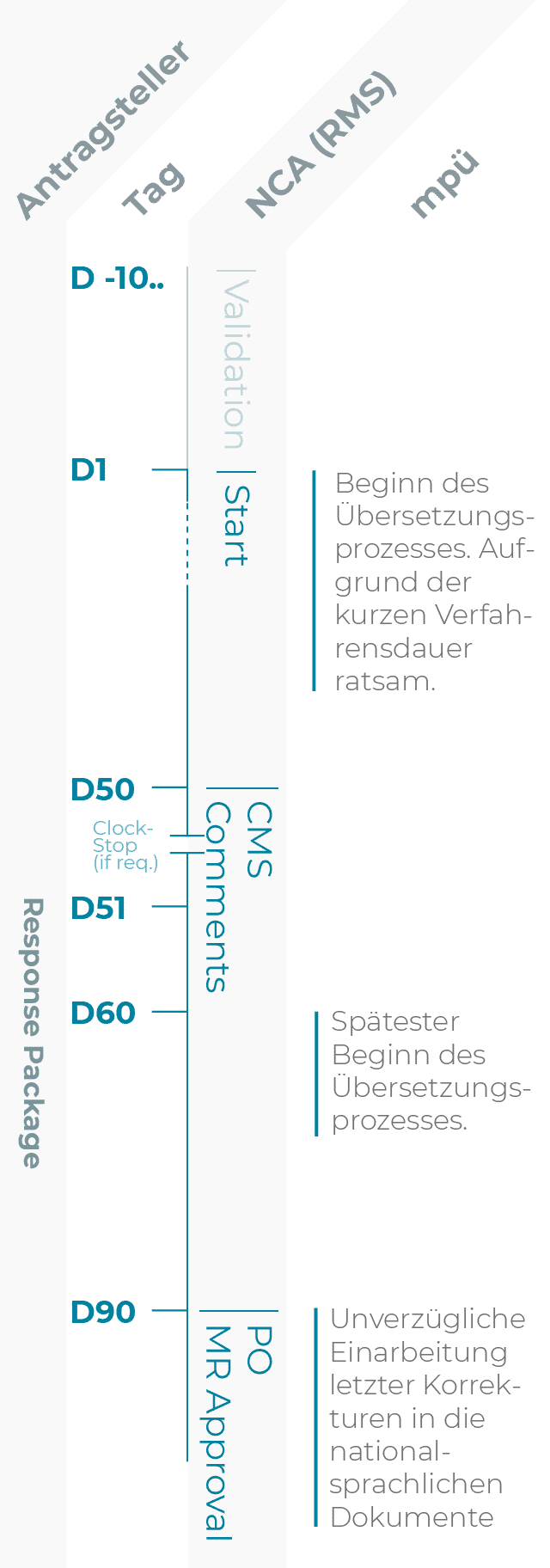
MRP-Verfahren
Die Verfahrensdauer beträgt 90 Tage.
Voraussetzung: Das betreffende Arzneimittel ist in mindestens einem Mitgliedstaat der EU zugelassen.
Vor Verfahrensstart
Das existierende Zulassungsdossier muss auf Aktualität geprüft und ggfs. durch Variations bei dem künftigen Reference Member State und ggfs. weiteren Mitgliedstaaten, in denen das betreffende Arzneimittel zugelassen ist, aktualisiert werden.
| D-10.. | Tag der Einreichung Der Antragsteller informiert mpü über die Einreichung des Zulassungsantrags und stellt die zu übersetzenden Dokumente zur Verfügung. Dies ermöglicht eine verlässliche Prozess- und Ressourcenplanung. Soweit sinnvoll, kann bereits mit den Übersetzungen begonnen werden. |
| D50 | Ab Tag 50, CLOCK STOP falls nötig Kommentare des CMS |
| D60 | Tag 60 Falls das Response Package Änderungen an Fachinformation (SmPC), Packungsbeilage (PIL) oder Labeling enthält, werden die geänderten Dokumente mpü mit Einreichung des Response Packages zur Verfügung gestellt. |
| D90 | Tag 90 Bei Konsens aller MS erteilt der RMS das MR-Approval. Nach dem MR-Approval sollten nur noch letzte Korrekturen durchgeführt werden. |
Immer pünktlich und jederzeit für Sie bereit
Einhaltung kurzer Zulassungsfristen
Um bei der Einreichung aller landessprachlichen Dokumente nach der „Positive Opinion“ (CP) bzw. nach dem „Approval“ (MRP/DCP) ein Versäumnis der Einreichungsfristen zu vermeiden, empfiehlt es sich, den Übersetzungsprozess der Quelltexte bzw. der Übertragung geänderter Textteile in die landessprachlichen Dokumente frühestmöglich zu beginnen,
d.h. optimalerweise ab dem Tag der Einreichung des Zulassungsantrages bzw. des Variation-Antrages/der Variation Notification.
Gemeinsam bringen wir Ihre Arzneimittel sicher und termingerecht auf den globalen Markt. Lassen Sie uns zusammenarbeiten.
info@mpue.com
+49 (0)731 954 950
Die 5-Tages-Frist nach Positive Opinion (CP) bzw. nach Approval (DCP/MRP) sollte letzten Änderungen vorbehalten sein.
Wochenendarbeit: kein Problem bei mpü.
Beim zentralen Zulassungsverfahren (CP) sind wir nach der Positive Opinion für die 5‑Tages‑Frist auch übers Wochenende für Sie im Einsatz, um alle Sprachversionen rechtzeitig zu finalisieren.
Anpassungen an die aktuellen QRD-Template-Versionen und Reviews/Lektorate bestehender Übersetzungen können schon vor Abschluss der Antragsprüfung bzw. vor Eingang der „Positive Opinion“ (CP) bzw. des „Approval“ (DCP/MRP) vorbereitend erfolgen.

Unsere schnelle Reaktionszeit sichert Ihre Deadline
Übersetzung aller Produktinformationen
Die Produktinformationen werden üblicherweise zunächst in Englisch verfasst und anschließend in alle relevanten EU-Sprachen übersetzt. Sie umfassen:
SmPC, PIL und Labeling
auch bekannt als:
- Fachinformation, Zusammenfassung der Merkmale des Arzneimittels, Summary of Product Characteristics (SmPC),
- Packungsbeilage, Gebrauchsinformation, Package Information Leaflet (PIL), Package Leaflet,
- Etikettierung, Etikett(en), Labeling, Text on Inner- and Outer Packaging
Life Cycle Management
Verfahren
- Zentrales Zulassungsverfahren (CP), auch beschleunigtes Verfahren (accelerated procedure)
- Dezentrales Zulassungsverfahren (DCP)
- Verfahren der gegenseitigen Anerkennung (MRP)
- Nationales Zulassungsverfahren (NP)
- Variations: Typ IA, Typ IB, Typ II
- Line Extensions
- Renewals
- Workshare
- Urgent Safety Restriction
- Annual Re-Assessment
- Article 61 (3) notifications (Label/PL/PIL)
- Referral procedure
- PSUR/PSUSAR/PBRER
Regularien
- QRD-Templates inkl. Anhänge
- Templates der nationalen Zulassungsbehörden
- EDQM Standard Terms
- Excipients Annex
- Annex A, Annex IV, Article 127a
- Table of non-standard abbreviations
- Names of EU-EEA countries
- Excipients Guide
- Compilation of QRD decisions on stylistic matters in product information
- Abbreviation of names of days on calendarised blisters
- QRD Convention
- Erstellung der PDF-Dateien: EMA PDF-User-Guide and -Checklist
- Länderspezifische Anpassungen wie Blue Box Requirements
- Synchronisierung/Erstellung der Unterlagen für verschiedene Produktstärken und Darreichungsformen
- Erstellung von Member State Versionen
Dokumente
- CTD/eCTD (Modules 1–5)
- GMP
- SOPs
- Korrespondenz mit Zulassungsbehörden
Korrekturzyklus mit Landesniederlassungen
Review durch die Landesniederlassungen des Kunden:
Management der Reviews und Durchführung der Korrekturen.
Übernahme des Linguistic Reviews mit Member States
- Etablierte Kontakte zu Ansprechpartner der Behörden aller EU-Member States
- Übernahme der gesamten Kommunikation zwischen Behörde und Übersetzer
- Inhaltliche und sprachliche Bearbeitung des Behördenfeedback
- Durch medizinisches und regulatorisches Know-how schnelle Problemlösung bei allen Rückfragen und Finalisierung der Labelings (Formblatt QRD 2)

Wir stehen für höchste Qualität seit über 45 Jahren.
Zulassungsdossier
Vorbereitung und Übersetzungen aller 5 Module des Common Technical Document (CTD)
Die Regelungen für die behördliche Genehmigung von Arzneimitteln sind in Europa weitgehend harmonisiert. Als Basis für die Arzneimittelzulassung am europäischen Markt dient das Common Technical Document (CTD bzw. eCTD).
Es enthält neben administrativen Informationen detaillierte Angaben zur pharmazeutischen Qualität und Herstellung, Wirksamkeit und Unbedenklichkeit des Arzneimittels (Nachweis durch Dokumentation klinischer und nicht-klinischer Studien).
Unser Prozess umfasst alle Schritte zur problemlosen Einreichung sowohl für das ganze Dossier als auch für einzelne Dokumente oder Module.
Format-Vorbereitung und -Finalisierung Ihrer Dokumente
Unabhängig vom Format der vorliegenden Ausgangsdokumente (z. B. Papierform, gescannte PDF, PDF-Dateien) können alle Vorlagen in elektronisch bearbeitbare Word-Formate zur Übersetzung konvertiert werden.
Die Übersetzungen werden entsprechend dem Layout der Ausgangstexte formatiert, inkl. Grafiken, Tabellen, etc..
Fachübersetzung in die gewünschten Sprachen
Da die einzelnen Module Inhalte unterschiedlicher fachspezifischer Bereiche umfassen, werden alle Dokumente den entsprechenden muttersprachlichen Fachübersetzern/-experten (z. B. Pharmazeuten, Mediziner, Molekularmediziner, Biochemiker, Biologen, Chemiker, Pharmakologen, Toxikologen) zur Übersetzung zugewiesen.
Qualitätssicherung durch Revision der Fachübersetzung nach DIN EN ISO 17100
Als erster Qualitätssicherungsschritt erfolgt die Revision durch einen weiteren Fachexperten. Ergänzt wird die Überprüfung durch Einsatz von speziell entwickelten Softwaretools zur Prüfung von Qualität und Konsistenz (Vollständigkeit, Zahlen, Produktnamen, chemische Substanzen, Abkürzungen etc.).
Labeling-spezifische Qualitätskontrolle in Modul 1
Dieser Schritt beinhaltet die Prüfung auf Einhaltung aller EMA-Regularien.
Erstellung des Dossiers im CTD-/eCTD-Format (auch ohne Übersetzung buchbar)
Wir prüfen alle Dokumente und erstellen das Zulassungsdossier im europaweit gültigen CTD-/eCTD-Format, unter Einhaltung aller regulatorischen Anforderungen für eine valide Antragstellung.
Durch die langjährige Erfahrung von mpü in der Partnerschaft mit orangeglobal können wir Ihnen ein ganzheitliches Leistungspaket aus Übersetzungen sowie Planung und Durchführung von Arzneimittelzulassungen anbieten.
Wir unterstützen Sie jederzeit bei der Einreichung von Variations und übernehmen für Sie gerne die erforderliche Umformatierung von Altformaten, z. B. aus dem NTA-Format in das CTD- bzw. eCTD-Format.
Zur Abbildung des aktuellen Dossierstandes gegenüber den europäischen und nationalen Behörden erstellen wir für Sie routinemäßig eine Baseline für Ihr Dossier.
Ihr Erfolg in der Arzneimittelzulassung beginnt mit den richtigen Übersetzungen.
Lassen Sie sich von uns beraten.

